UCSF ChimeraX é um software de visualização e análise estrutural de biomoléculas desenvolvido pela equipe da University of California. Ele é um software gratuito com suporte multiplataforma que pode ser adquirido em https://www.cgl.ucsf.edu/chimerax/index.html.
Neste tutorial, você aprenderá alguns dos comandos básicos do ChimeraX.
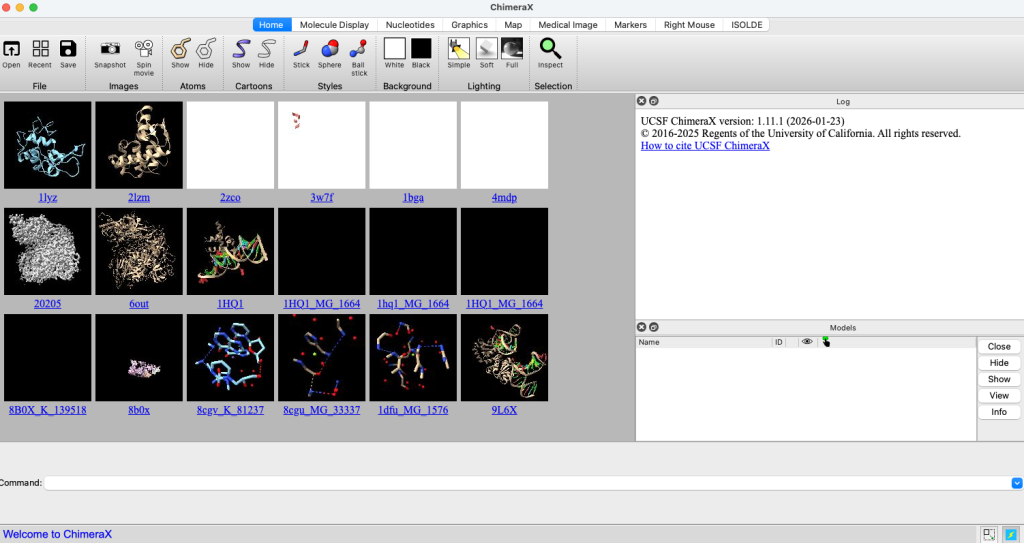
Carregando uma estrutura
Para carregar uma estrutura, você pode usar o comando open.
open 2lzmObserve que a estrutura da lisozima 2lzm é carregada na área principal da interface. No lado direito, é exibido um painel de log com informações sobre os comandos executados e mensagens do sistema. Logo abaixo, encontra-se a lista de modelos atualmente carregados.
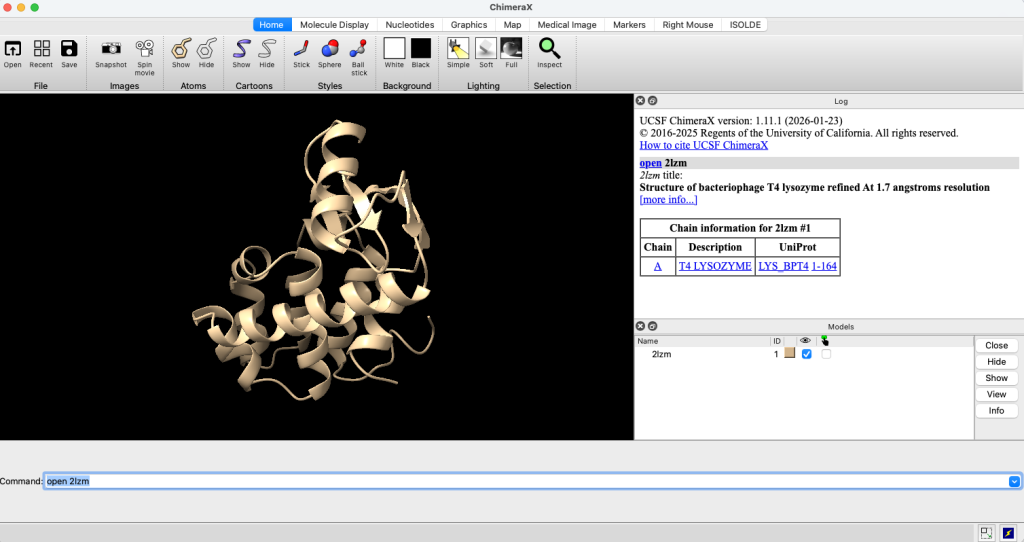
Clique no checkbox abaixo do olho para ocultar ou exibir a interface. Para selecioná-la, clique no checkbox ao lado.

Utilize o menu superior para alterar o estilo da visualização:
Excluir a estrutura
Você pode excluir a estrutura usando o comando close. Entretanto, para fechar a estrutura, precisamos do seu ID. Você pode consultar isso no painel de models:
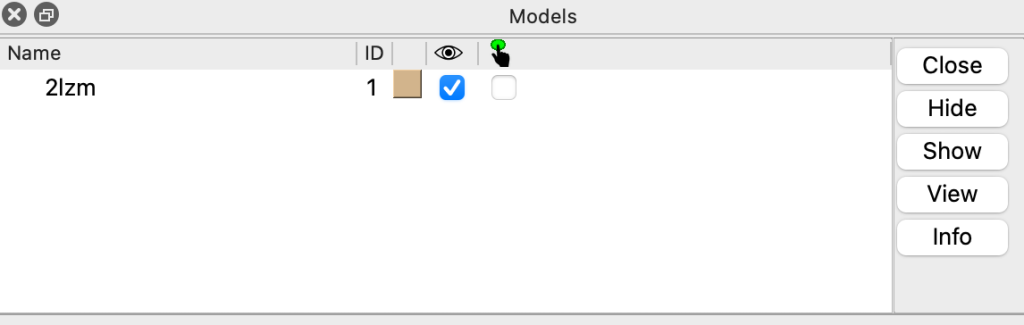
Opcionalmente, você pode usar o comando info models:

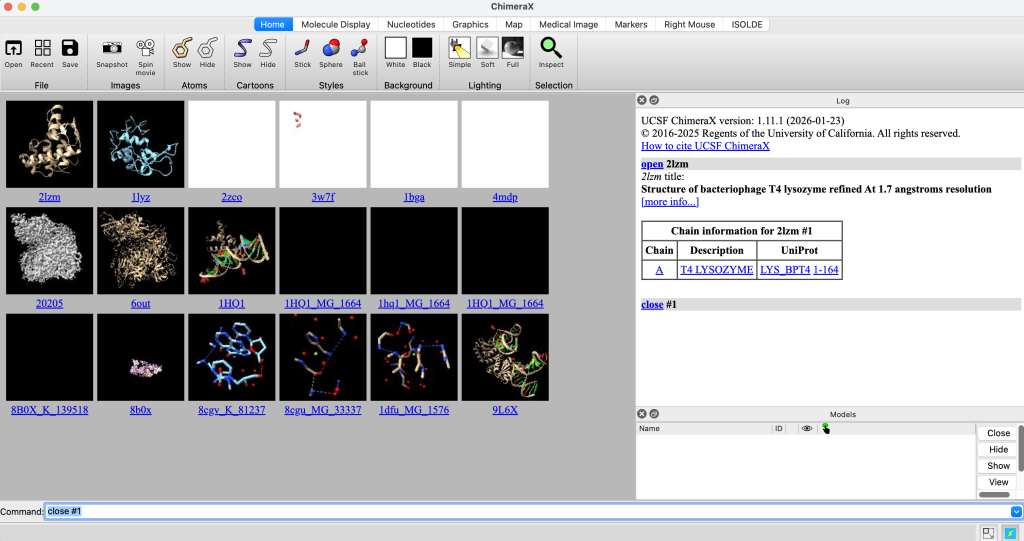
Agora que sabemos o ID, podemos excluí-lo com o comando:
close #1Observe que a estrutura foi excluída:
Alinhando estruturas
Agora, vamos abrir duas estruturas e alinhá-las. Vamos começar carregando as duas estruturas:
open 2lzm
open 1lyz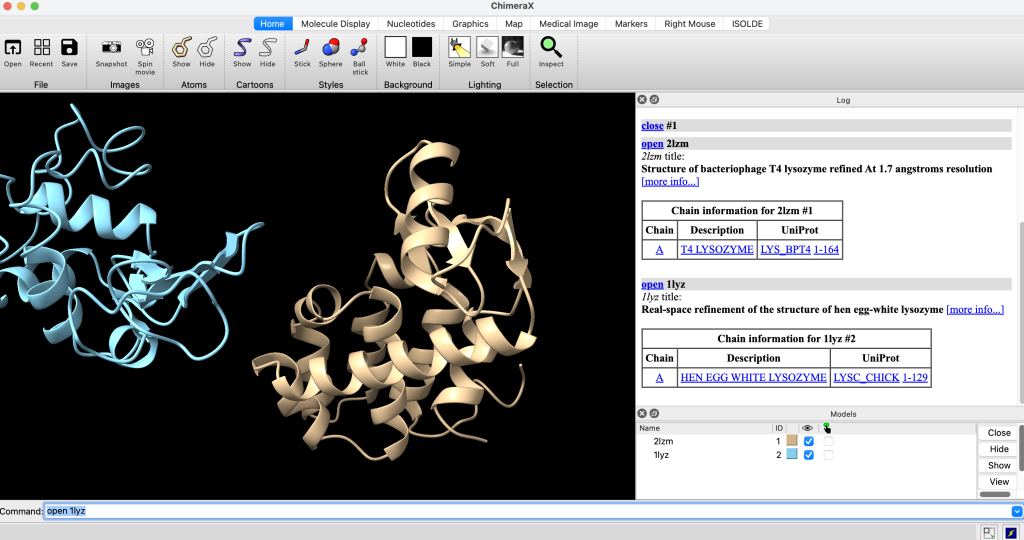
Agora vamos usar o comando MatchMaker (mm) para alinhar sequências, identificar resíduos equivalentes e fazer superposição estrutural.
No terminal de linhas de comandos execute:
mm #2 to #1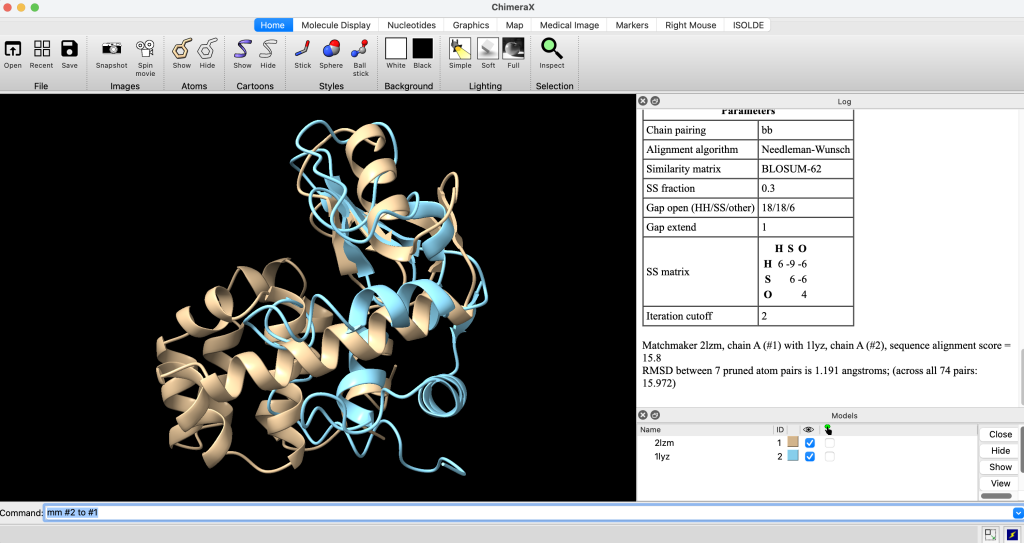
Caso você deseje ver o alinhamento, execute o comando:
mm #2 to #1 showAlignment true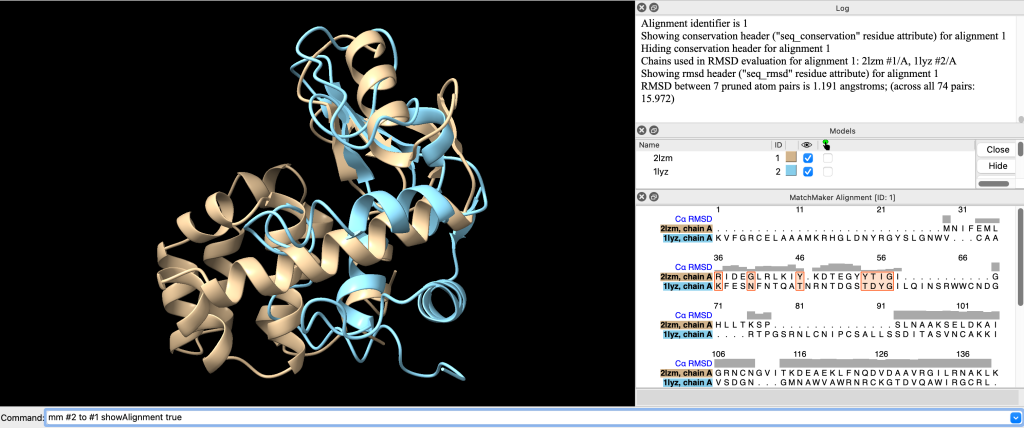
Para consultar o RMSD do alinhamento, habilite a opção verbose da seguinte forma:
mm #1 to #2 verbose trueObserve o resultado:
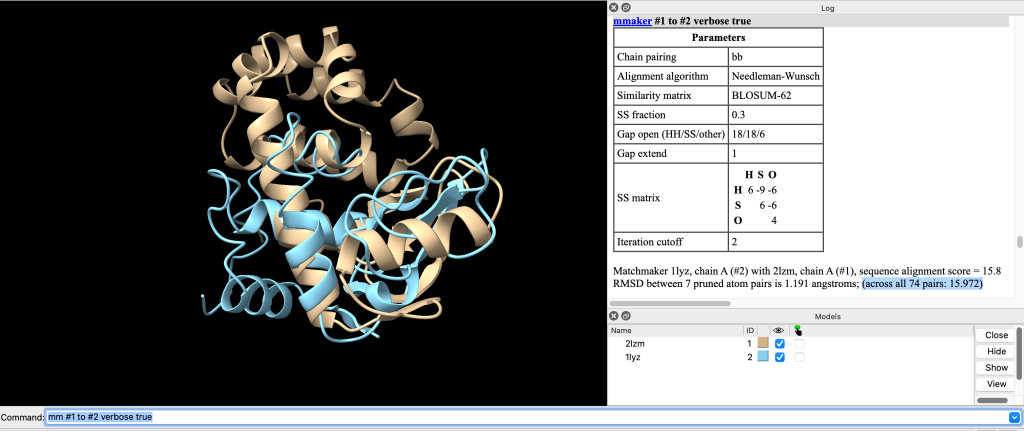
Observe que você pode executar o comando rmsd #1 to #2 para obter o rmsd, mas, neste caso, ele não funcionará, pois este comando exige que as estruturas tenham a mesma quantidade de átomos.
Selecionando partes de uma proteína
Agora vamos aprender a selecionar partes de uma proteína. Podemos selecionar parte de um modelo usando o comando select. O padrão é o seguinte:
select #ID/CADEIA:RESÍDUO@ÁTOMOPor exemplo, vamos selecionar uma cadeia. Vamos começar selecionando a cadeia B. Carregue a estrutura 1bga e, em seguida, selecione a cadeia B com o comando:
select /B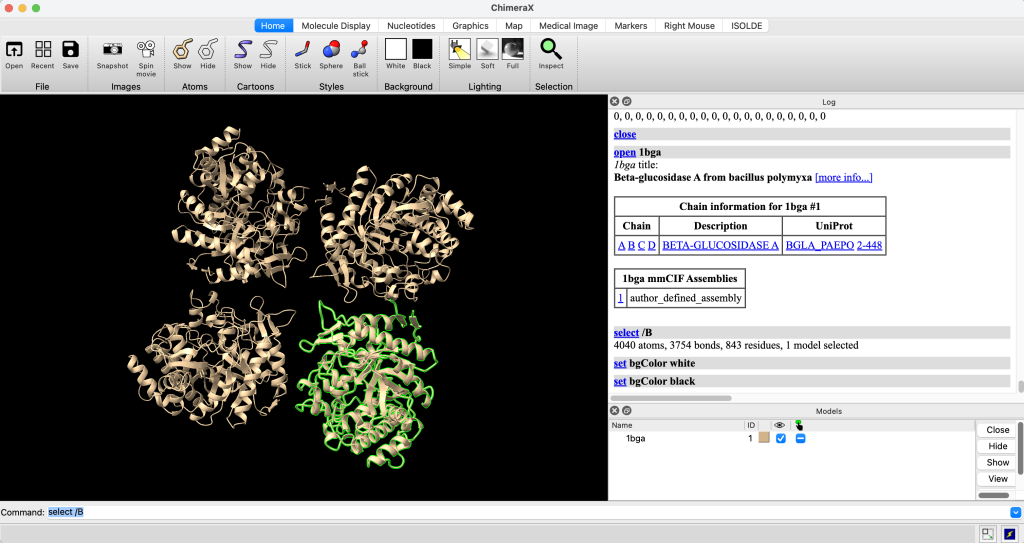
Você pode limpar a seleção usando:
select clearAgora, selecione novamente a cadeia B e vamos colorí-la de azul. Você pode fazer isso usando o comando:
color sel blue cartoonsObserve o resultado:
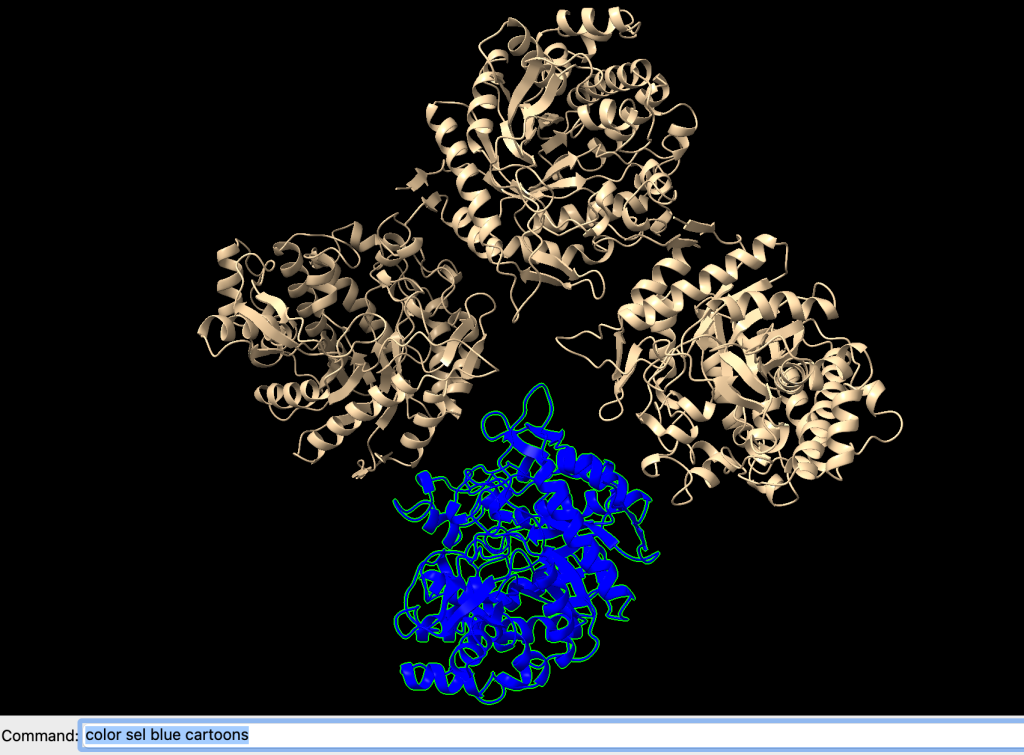
Este azul não ficou muito bom, então podemos testar outras variações de cores:
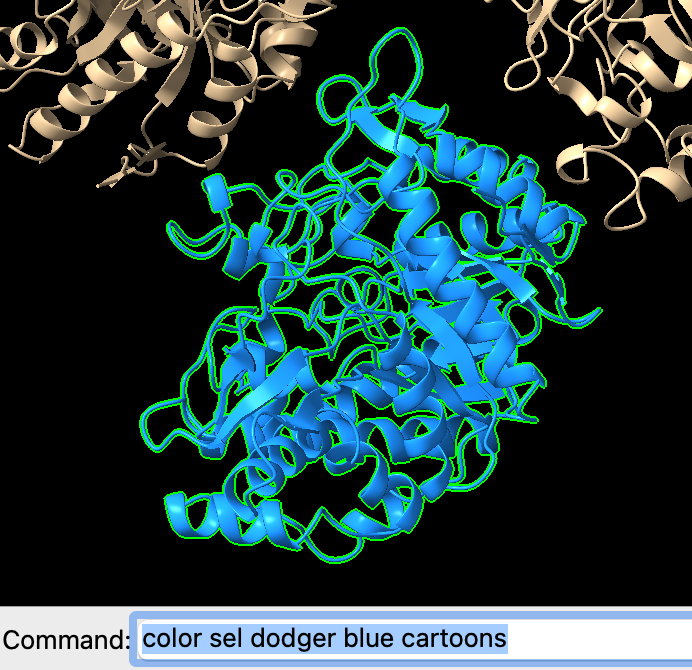
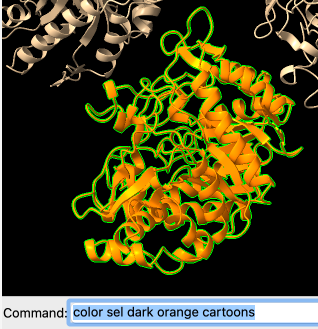
Deletando partes de uma estrutura
Agora vamos apagar a cadeia selecionada anteriormente usando o comando:
select /B
delete selAntes
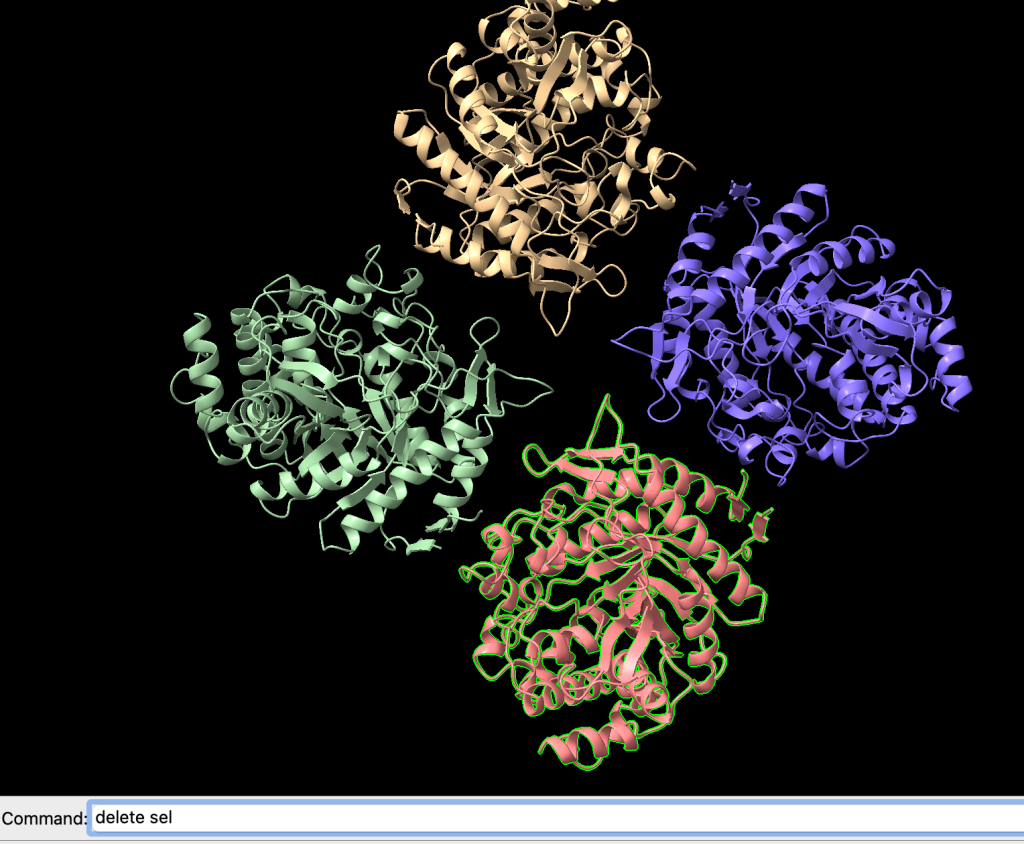
Depois
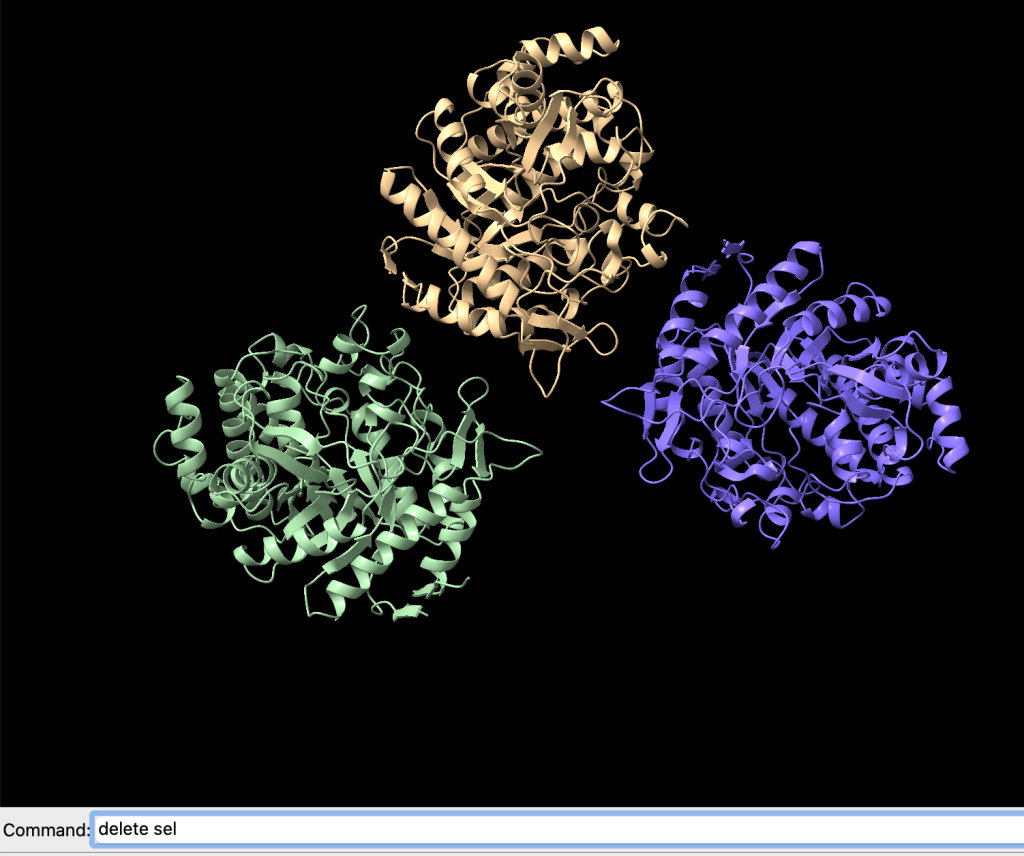
Você pode especificar exatamente a cadeia que deseja excluir sem precisar selecioná-la, da seguinte forma:
delete #ID/CADEIAPor exemplo, podemos excluir a cadeia C da seguinte forma:
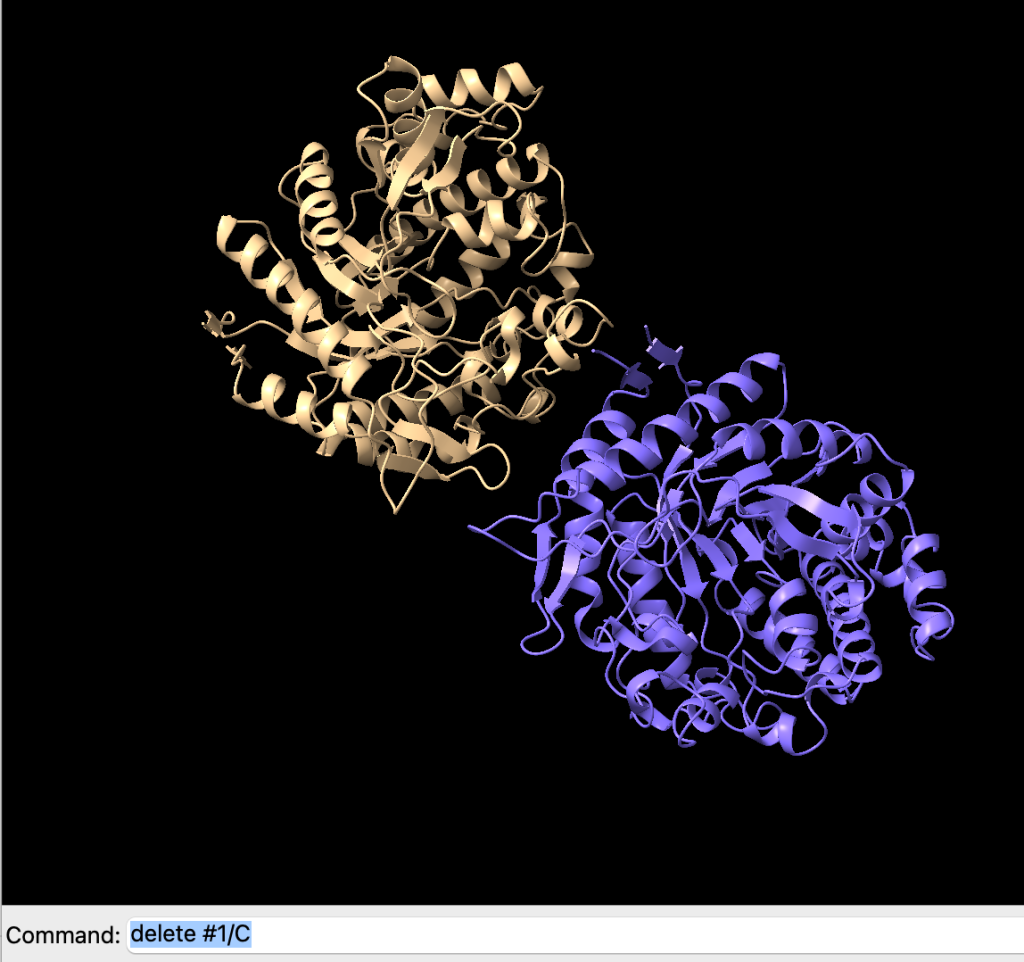
Carregando a sequência
Podemos exibir a sequência carregando o painel de sequências pelo menu Molecule Display > Sequence.
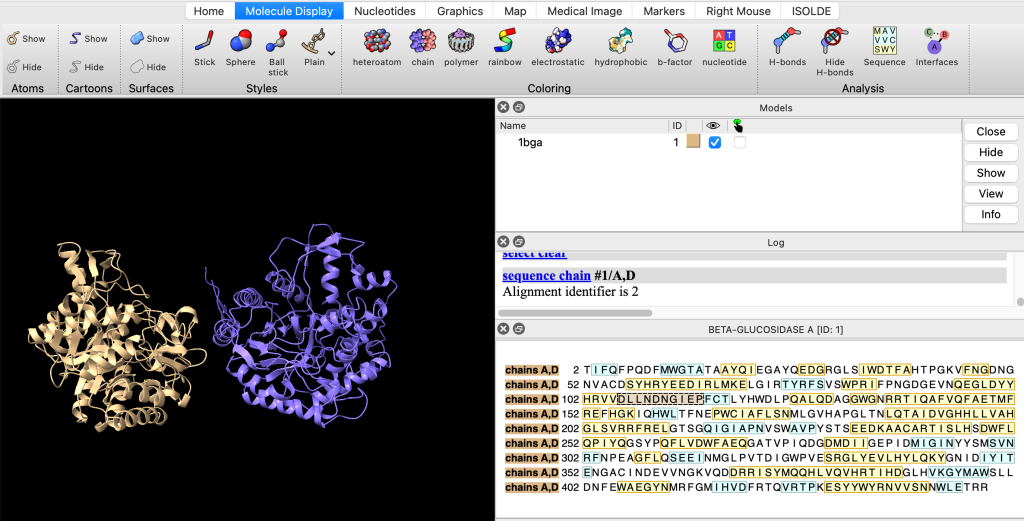
Observe que clicar em partes da sequência, seleciona as regiões especificadas:
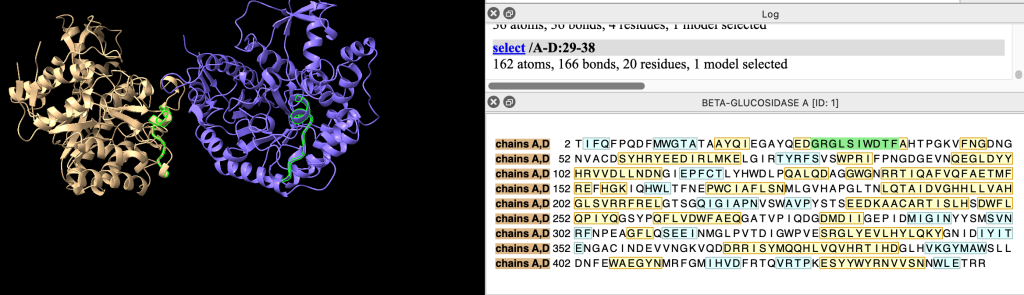
Centralizando a estrutura
Ao deletarmos a cadeia D de 1BGA, perceba que a única cadeia remanescente ficará desalinhada. Podemos recentralizá-la usando o comando:
view /A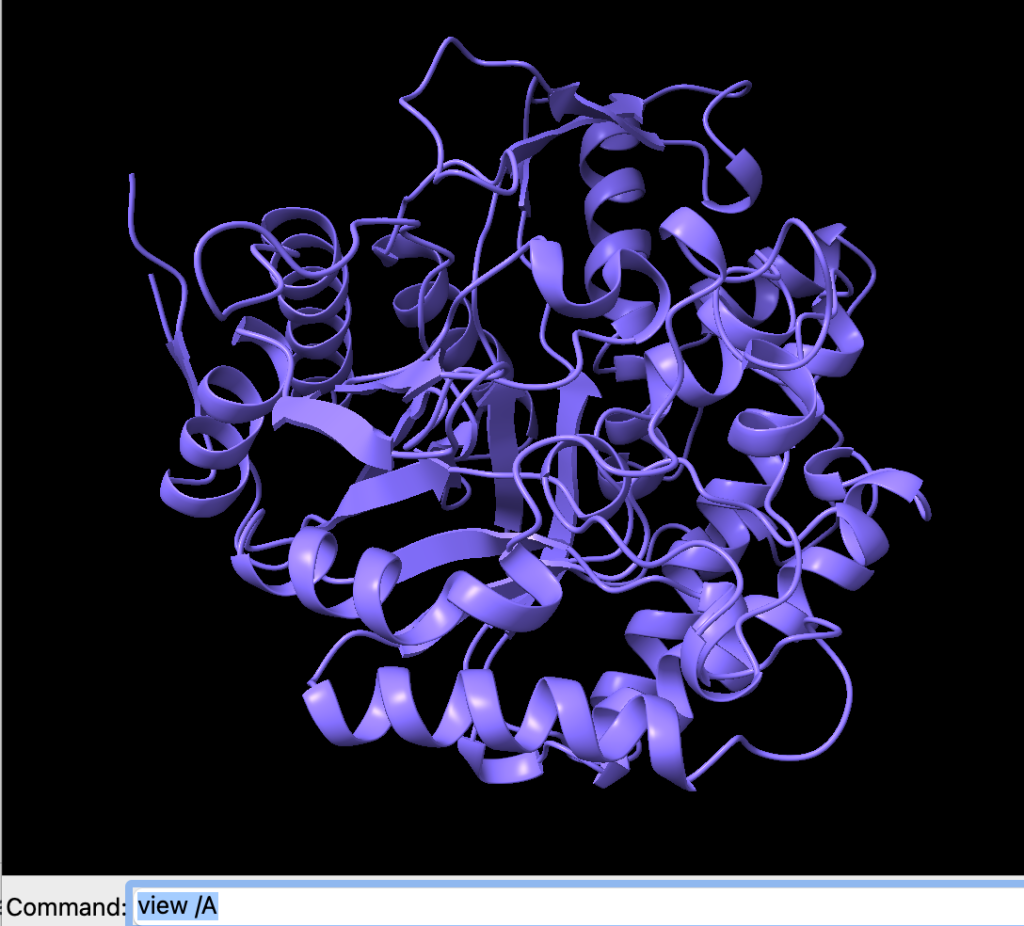
Selecionando o sítio de ligação
Agora, vamos selecionar os resíduos do sítio de ligação da cadeia A da beta-glicosidase 1BGA. O sítio ativo de 1BGA é composto por dois glutamatos (166 e 352). Podemos selecioná-los com o comando:
select #1/A:166,352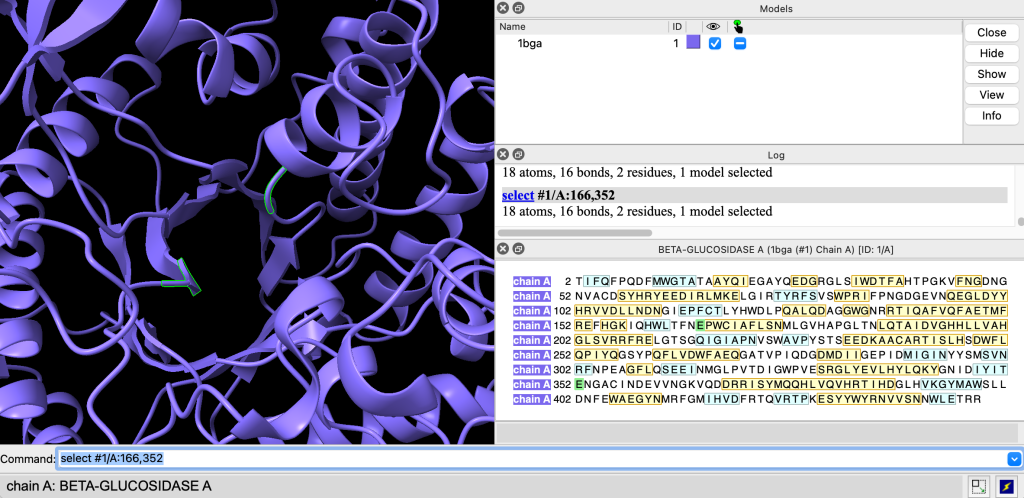
Agora exiba os átomos usando o comando:
show sel atoms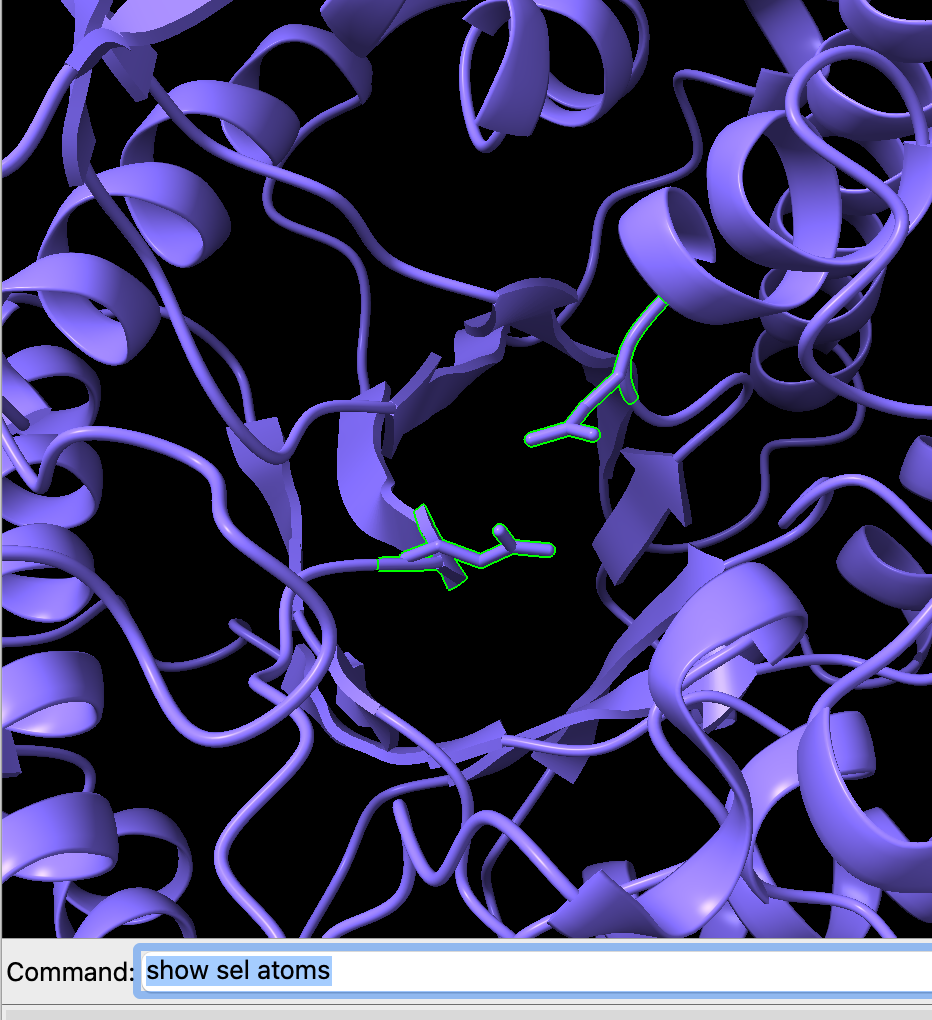
style sel stickAgora, vamos colorir os sticks de laranja. Podemos fazer isso usando o comando:
color sel dark orange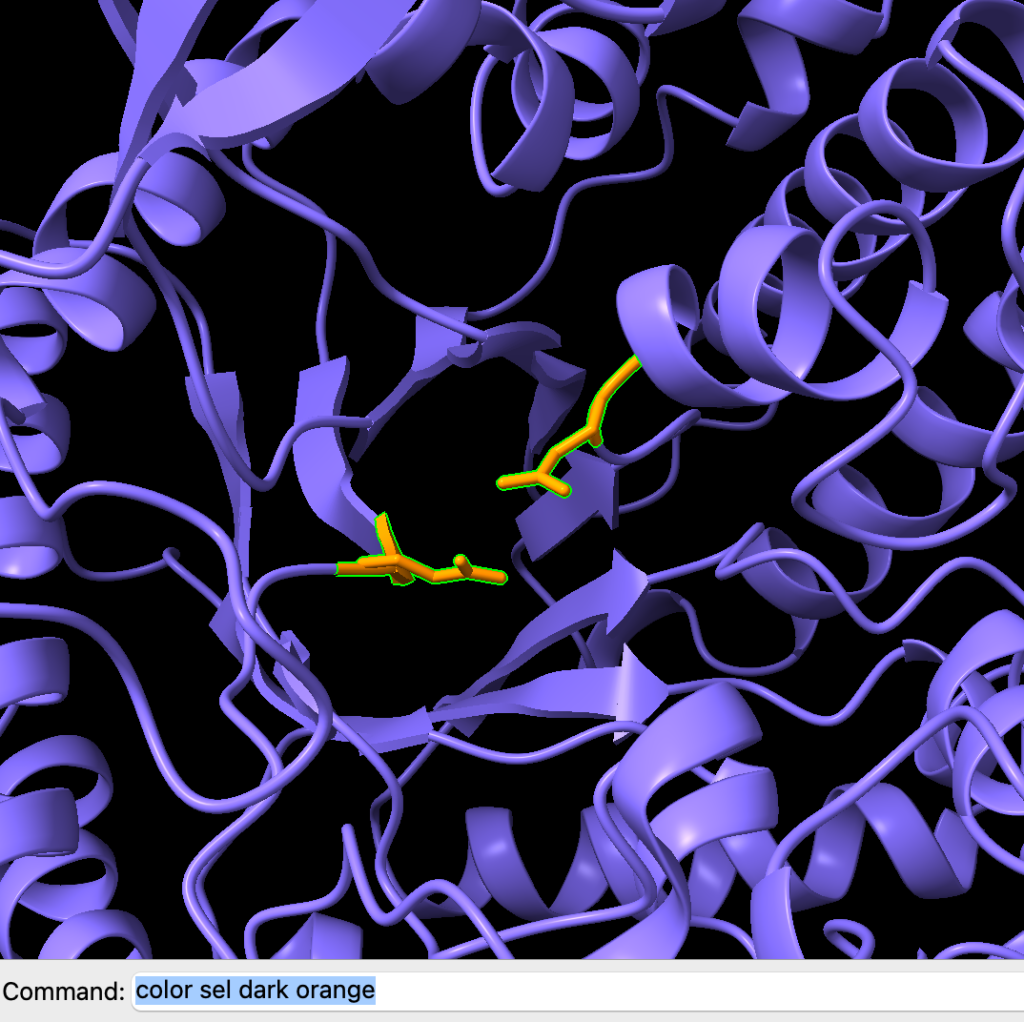
Agora, vamos alterar as cores dos átomos de oxigênio e nitrogênio. Podemos fazer isso com o comando:
color sel byhetero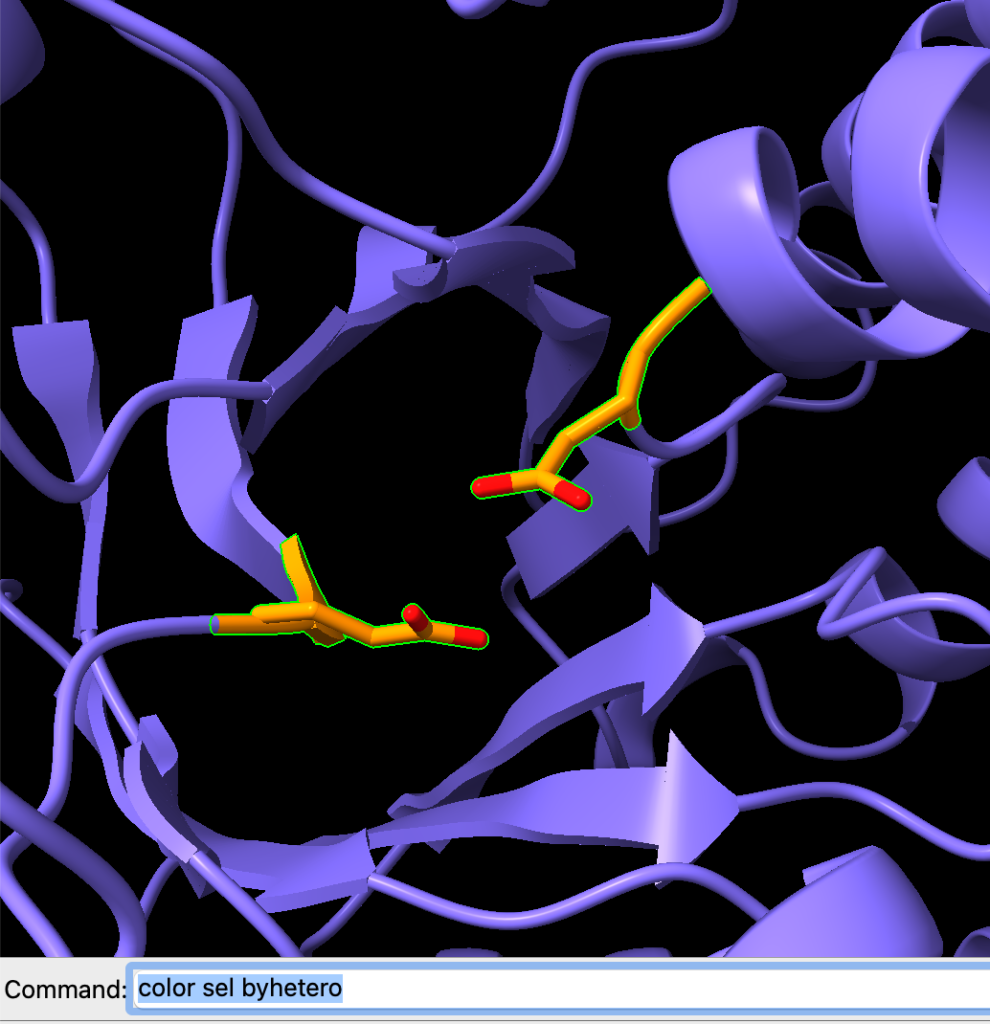
Podemos exibir as esferas usando o comando:
style sel ball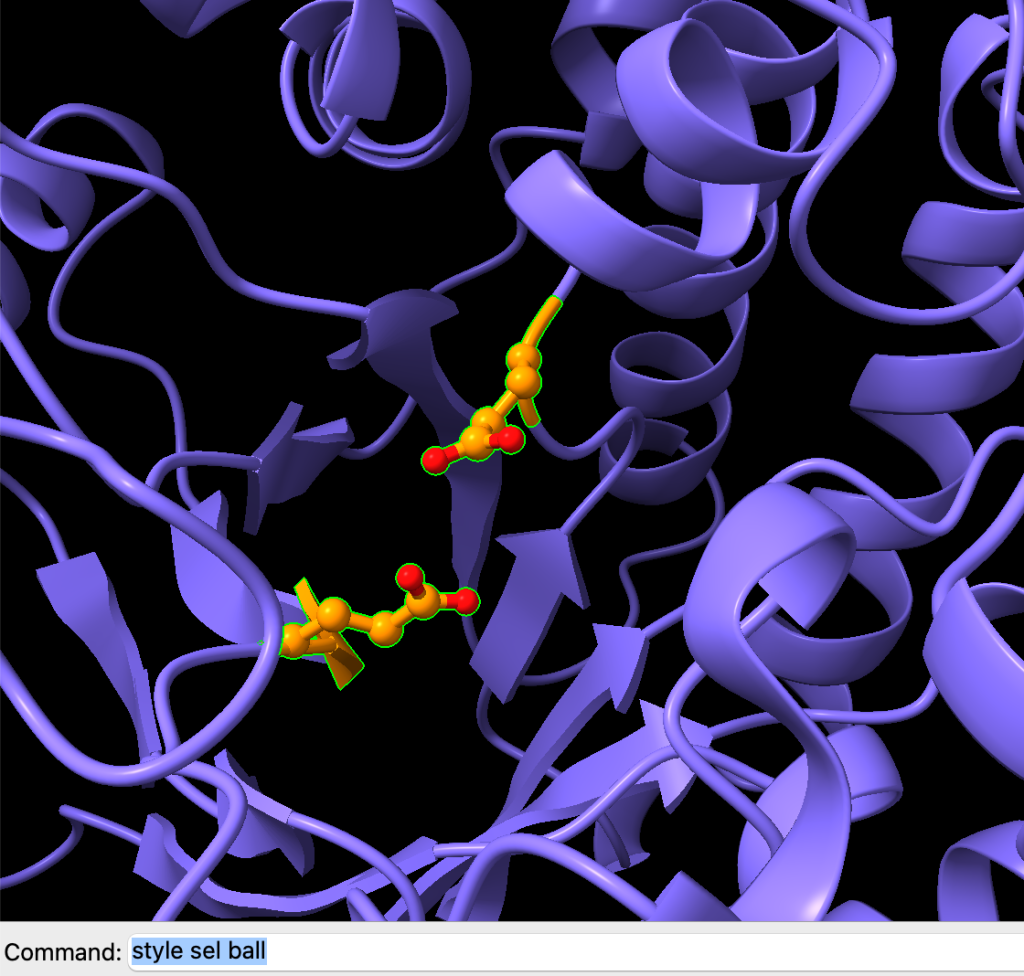
Selecionando uma região próxima ao sítio
Podemos selecionar todos os resíduos a uma distância de 6 Å dos resíduos catalíticos usando o comando:
select zone #1/A:166,352 6Observe que selecionamos 159 átomos:
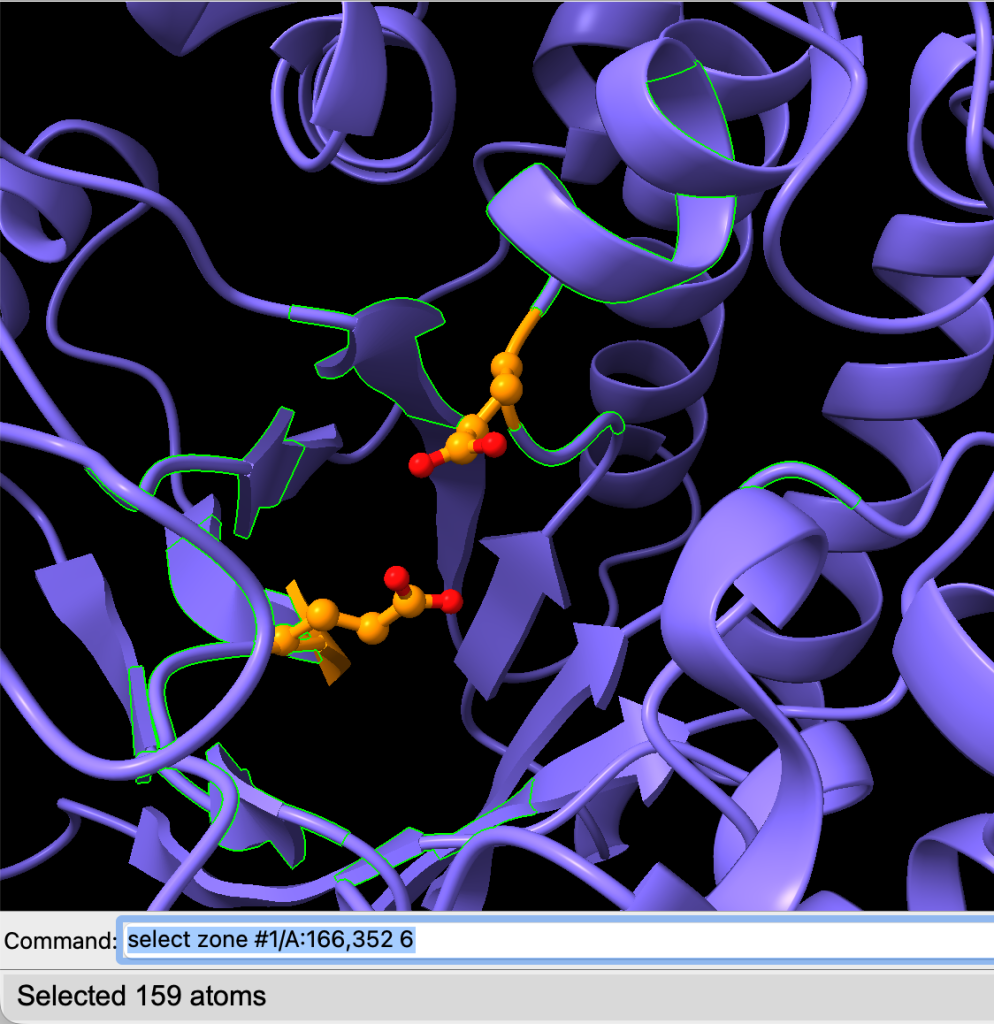
Agora, exiba os átomos usando o comando:
show sel atoms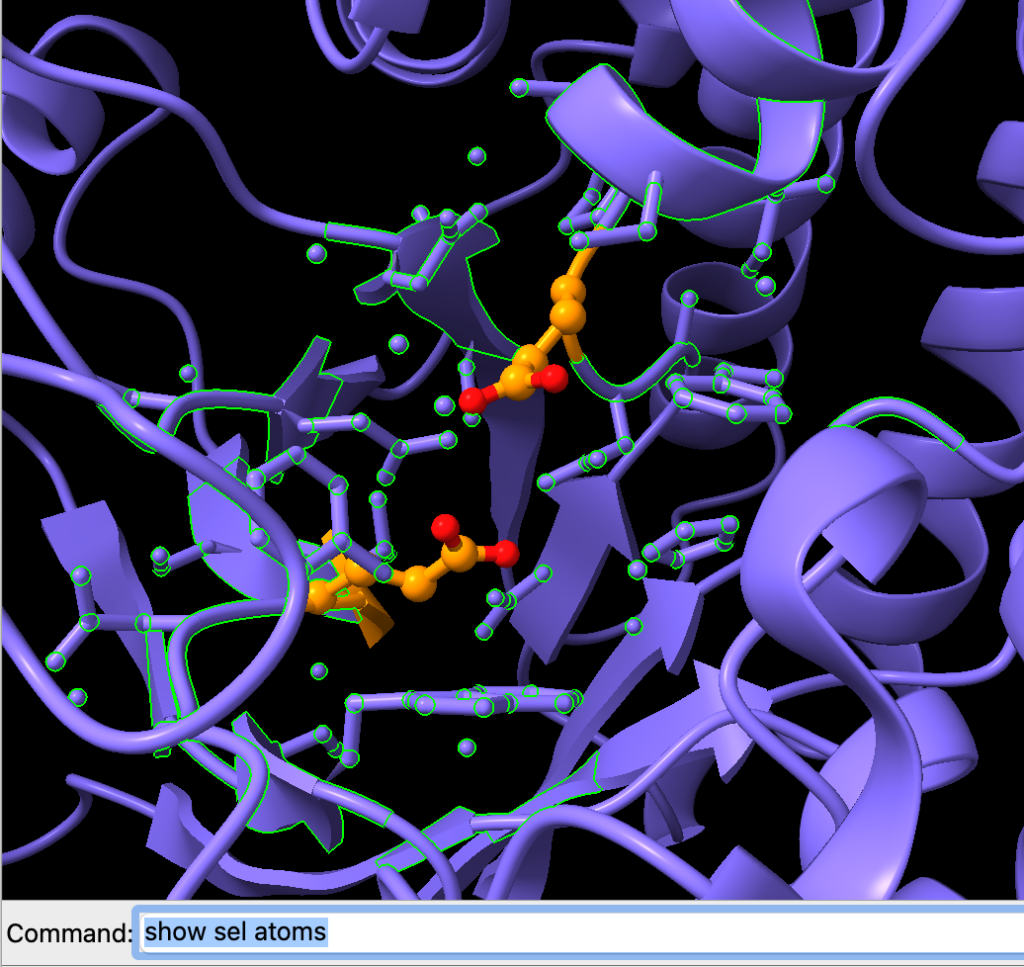
Vamos estender a seleção a todos os átomos dos resíduos da região. Use o comando:
select up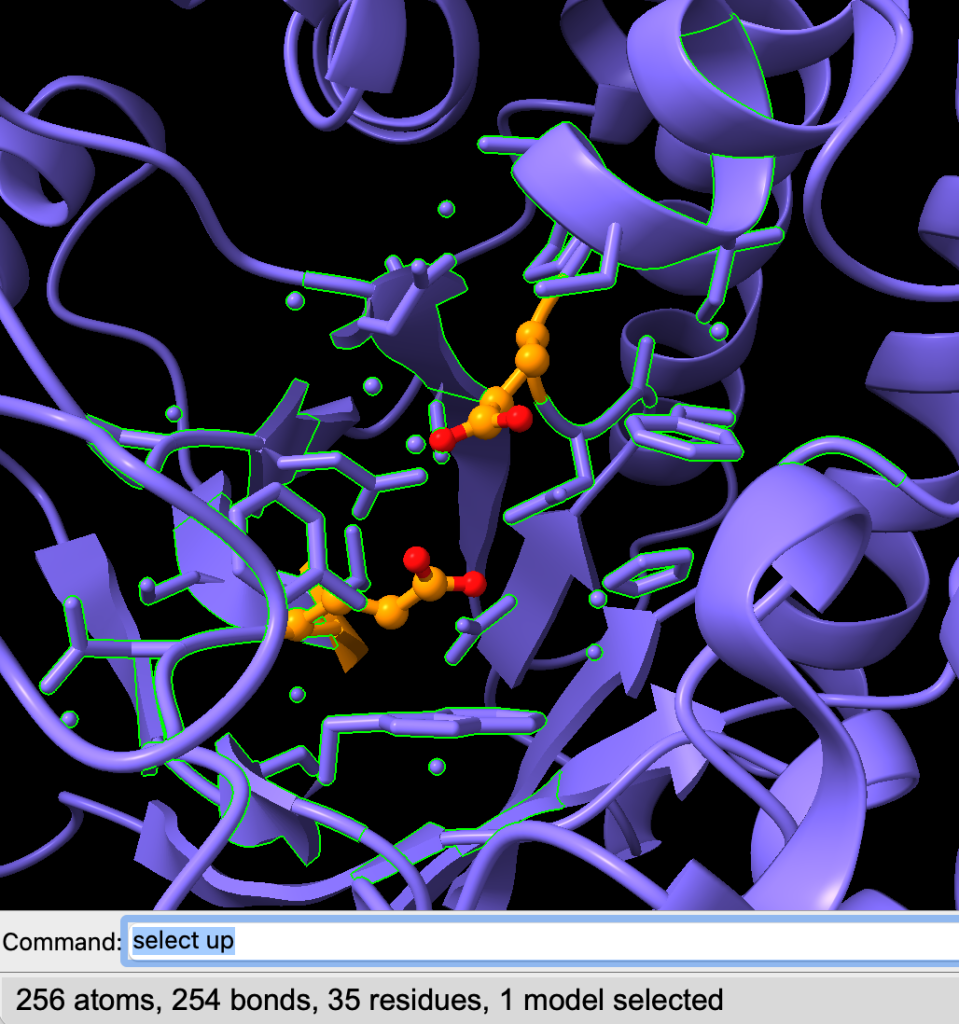
Agora vamos aplicar o esquema de cores por heteroátomos:
color sel byhetero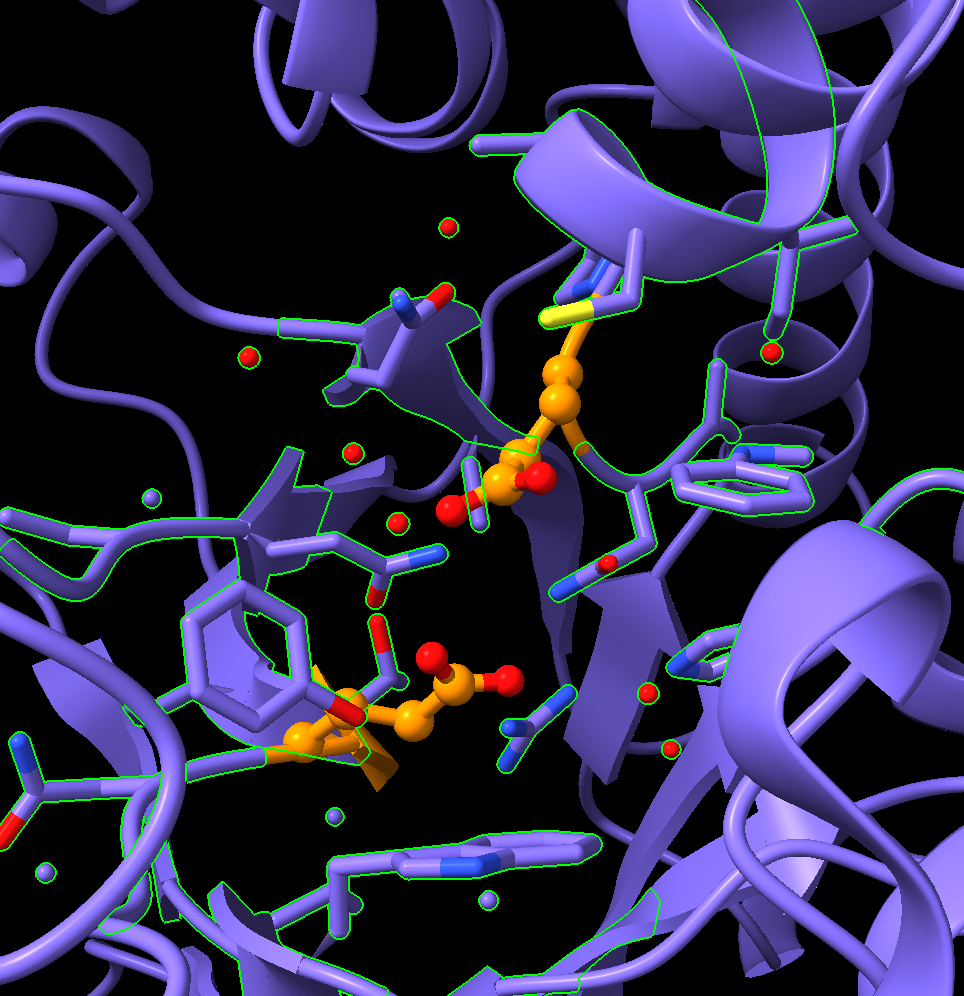
Note que ainda faltam alguns átomos. Vamos estender mais uma vez a seleção e exibir os átomos faltantes:
select up
show sel atoms
Dando zoom no sítio de ligação
Vamos limpar a seleção e depois dar um zoom no sítio de ligação.
select clear
view #1/A:166,352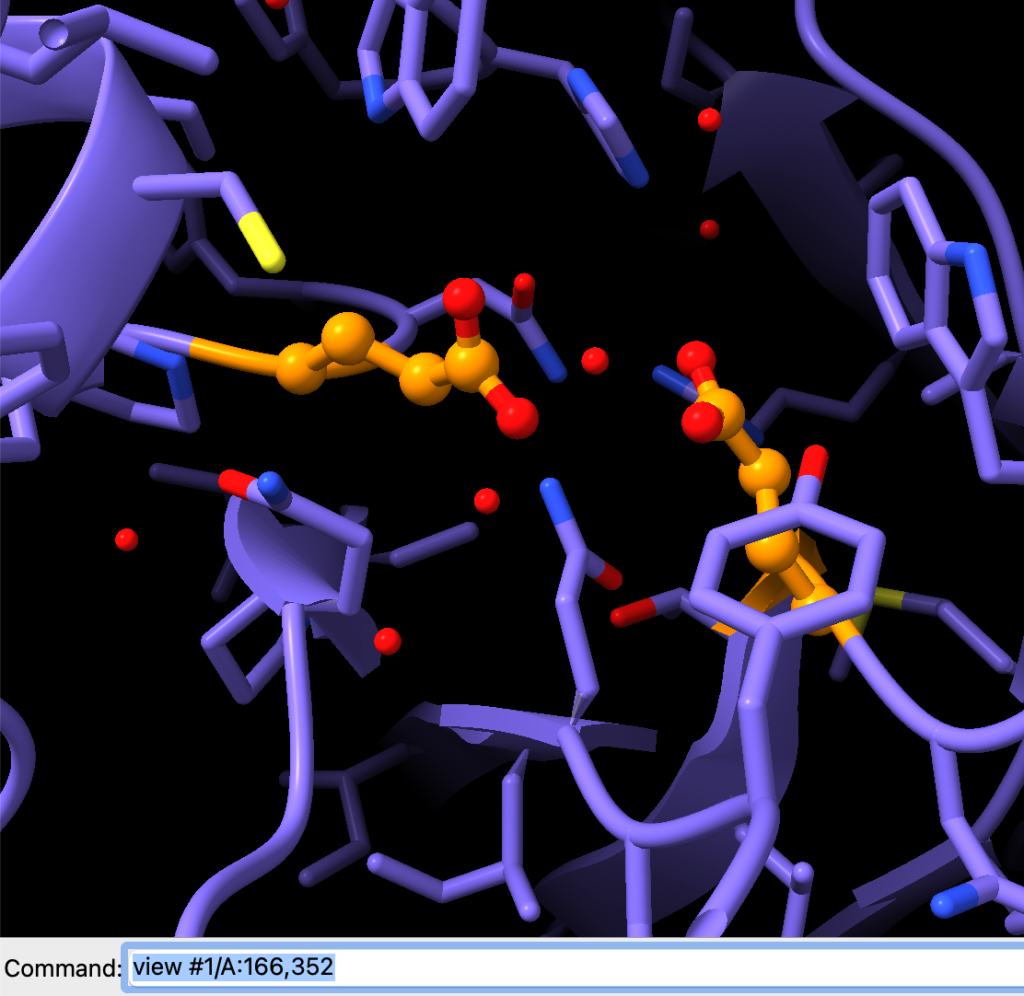
Medindo a distância entre dois átomos
Agora, vamos medir a distância entre dois átomos. Podemos fazer isso usando o comando distance seguido de dois átomos selecionados.
distance #1/A:352@OE1 #1/A:166@OE2Observe o resultado:
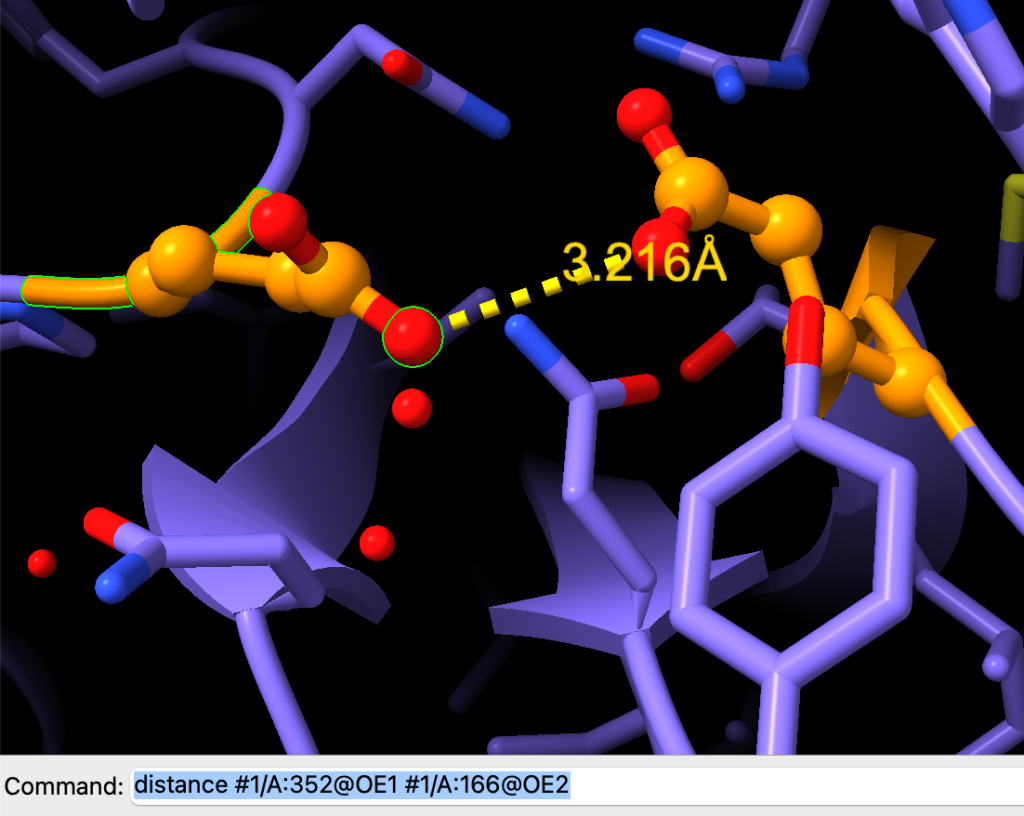
Podemos alterar o tamanho da fonte usando o comando line height.
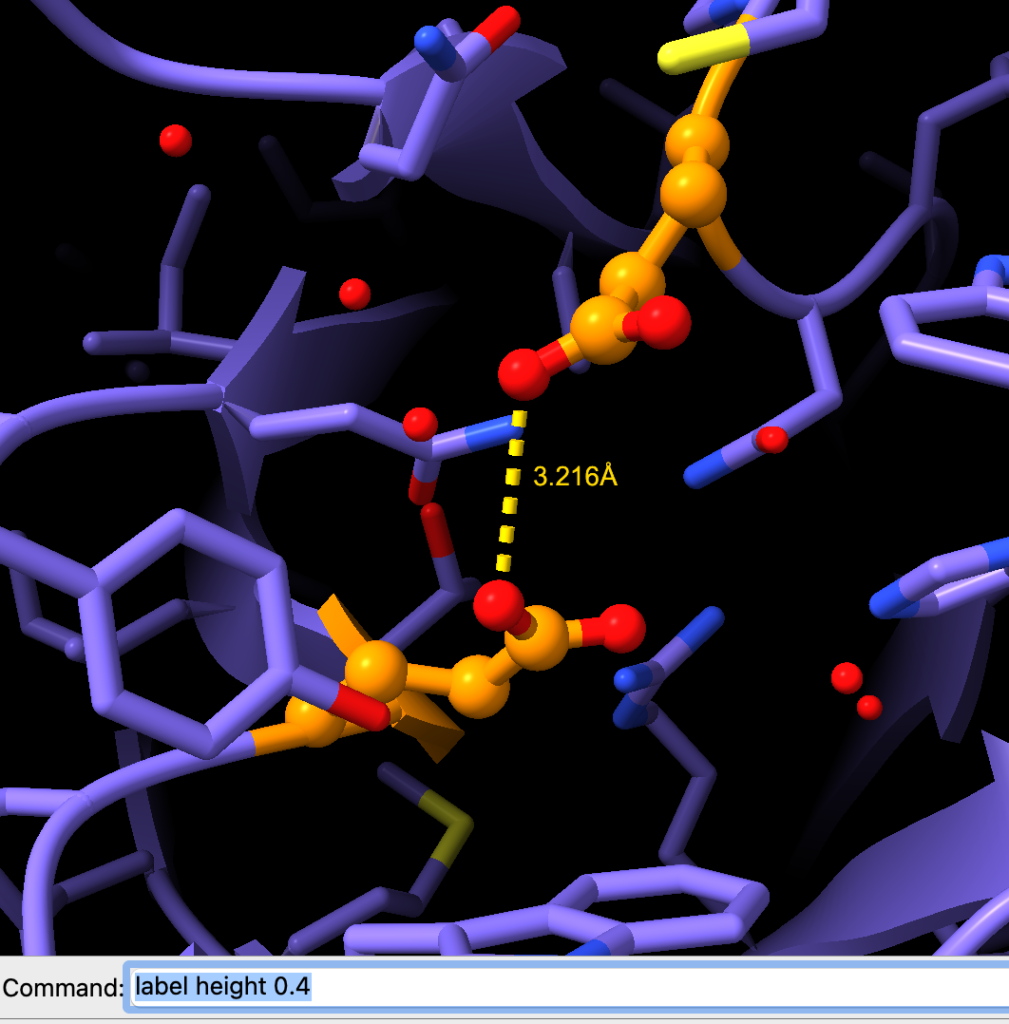
Em seguida, vamos alterar a cor de fundo para branco e, em seguida, alterar a cor da fonte para cinza:
set bgColor white
label color grey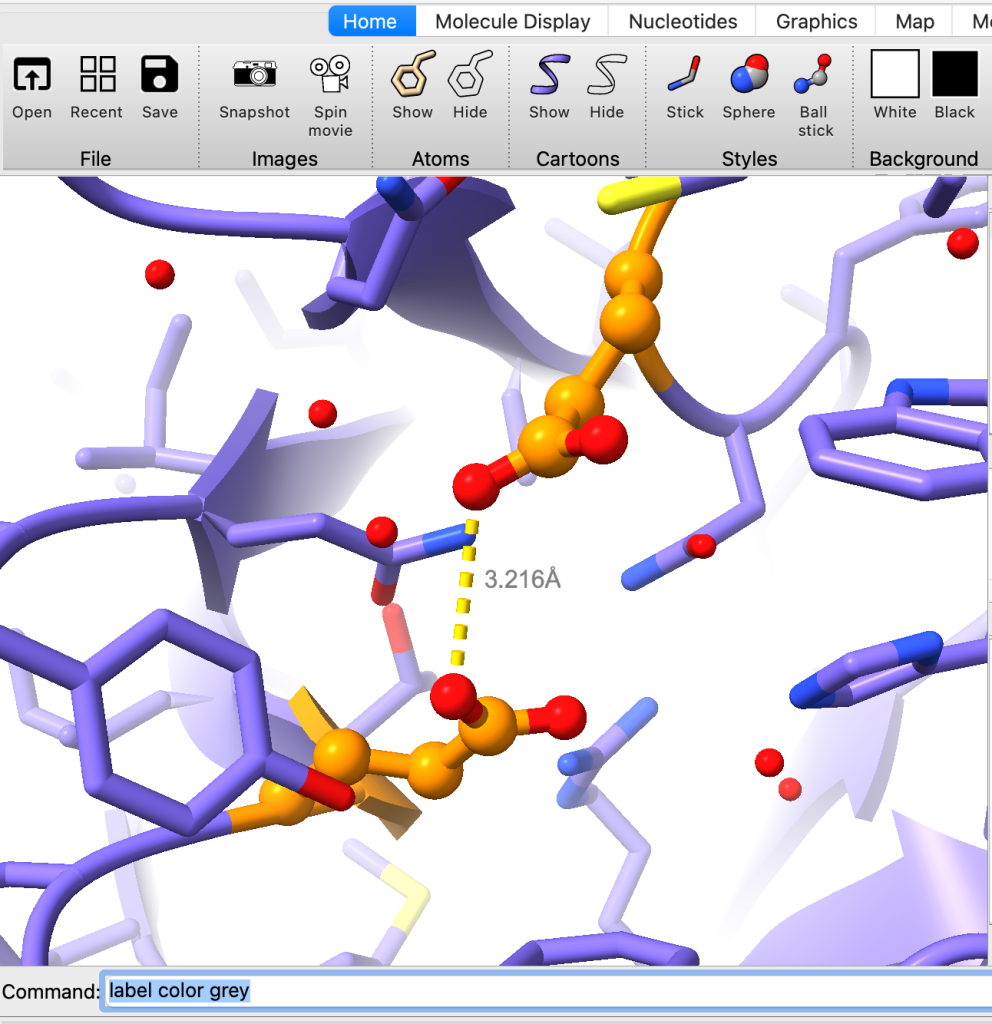
Como exibir rótulos dos resíduos
Agora, vamos adicionar rótulos (labels). Vamos começar com os dois glutamatos:
select #1/A:166,352
label sel residues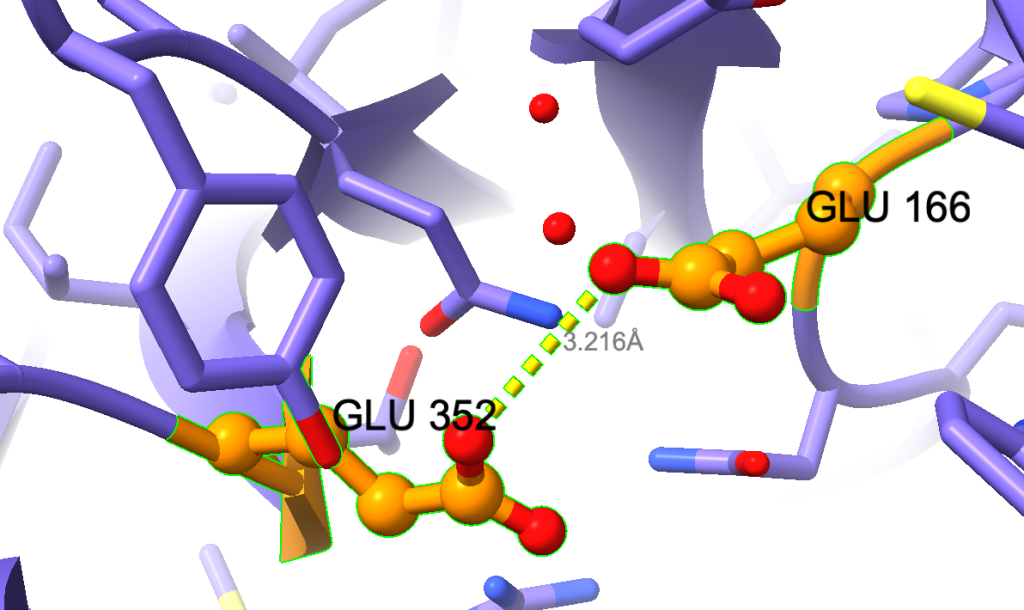
Você pode remover as labels usando:
label deleteou label delete sel para excluir apenas a seleção.
Infelizmente, isso apaga as distâncias que criamos anteriormente. Vamos refazê-las. No painel de modelos, selecione distances e depois clique no botão close.
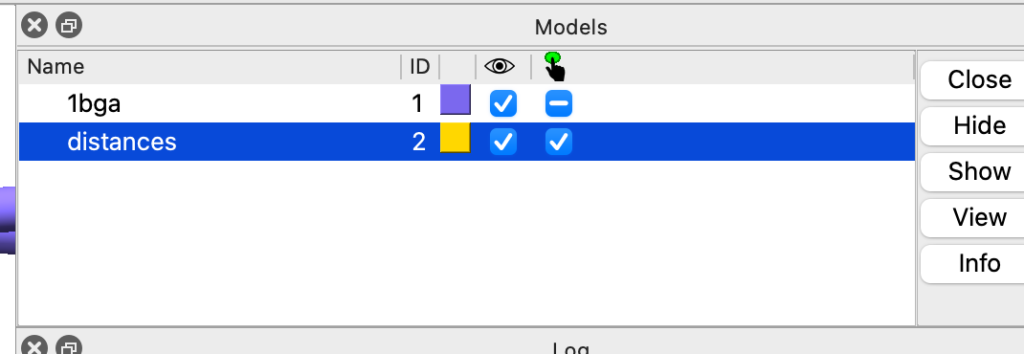
Adicione novamente as distâncias com os comandos:
distance #1/A:352@OE1 #1/A:166@OE2
label color grey
label height 0.4Agora adicione as labels dos resíduos usando:
label sel residues
label sel color blackVeja o resultado:
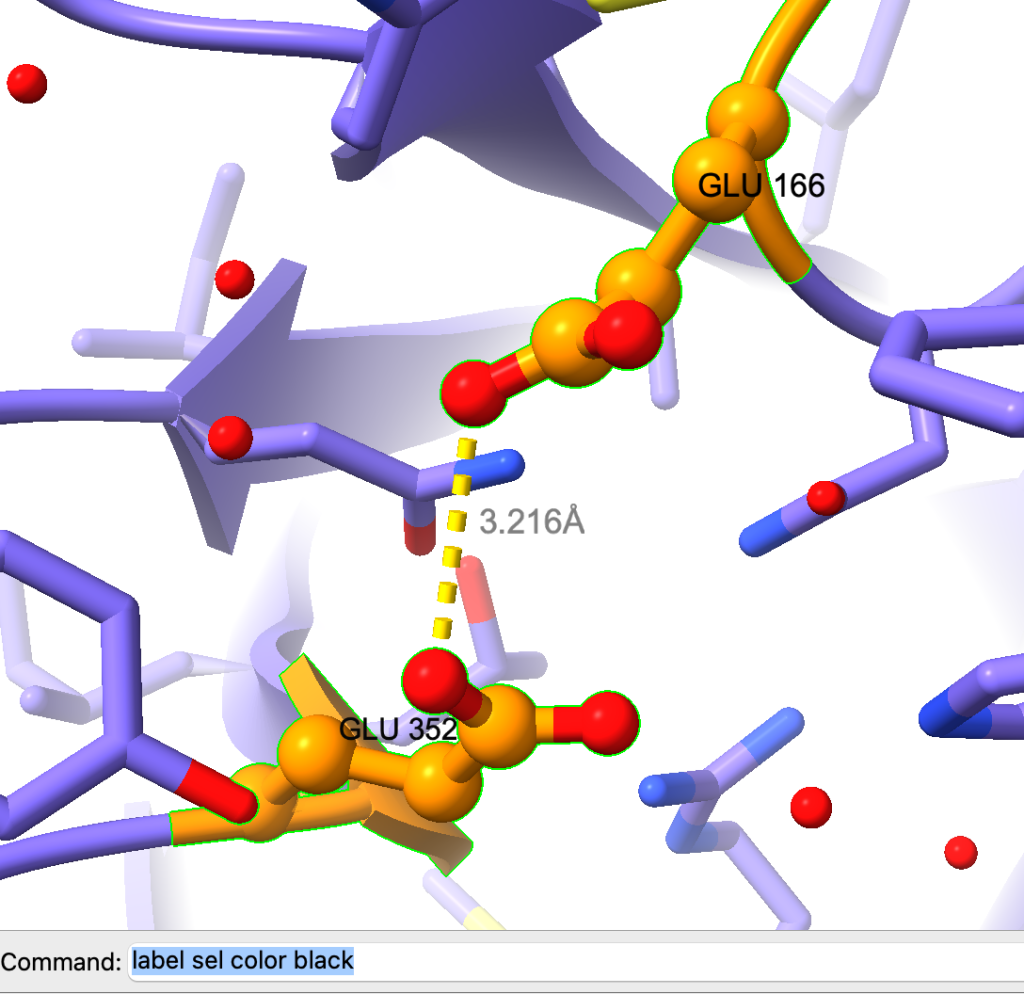
Podemos melhorar mais ainda o estilo das labels, veja como:
label sel color white
label sel height 0.3
label sel bgColor blackObserve o resultado:
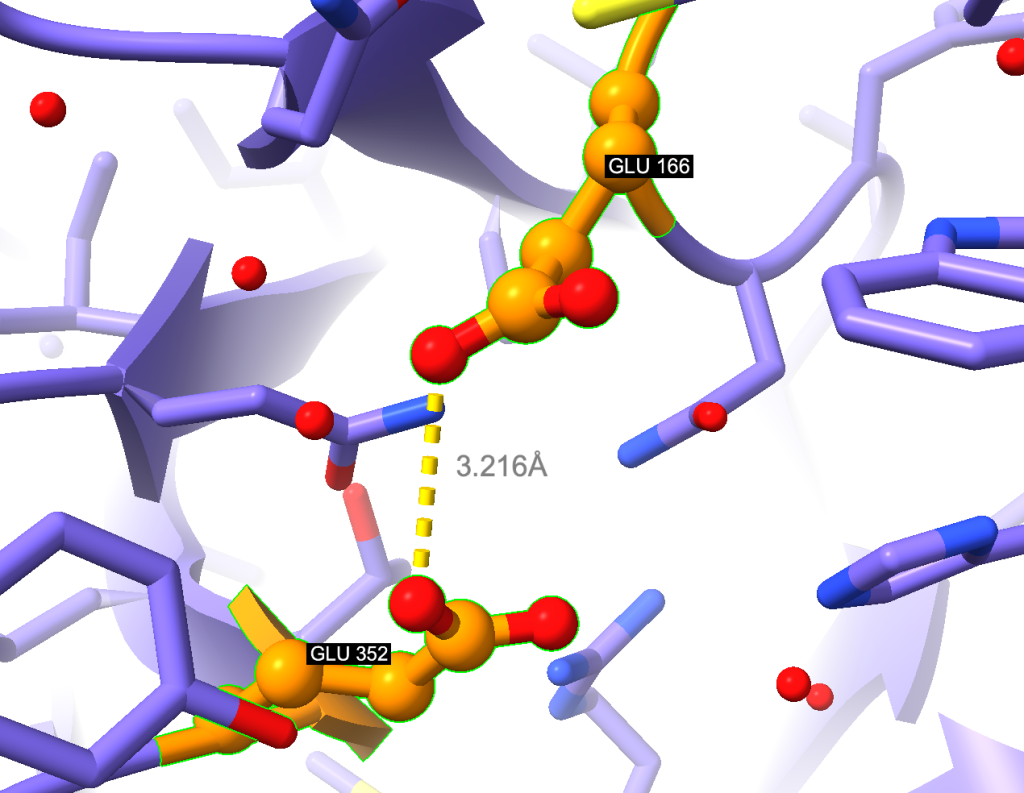
Adicionando superfície
Podemos adicionar superfície e deixá-la semitransparente usando os seguintes comandos:
surface
transparency 70 surfaces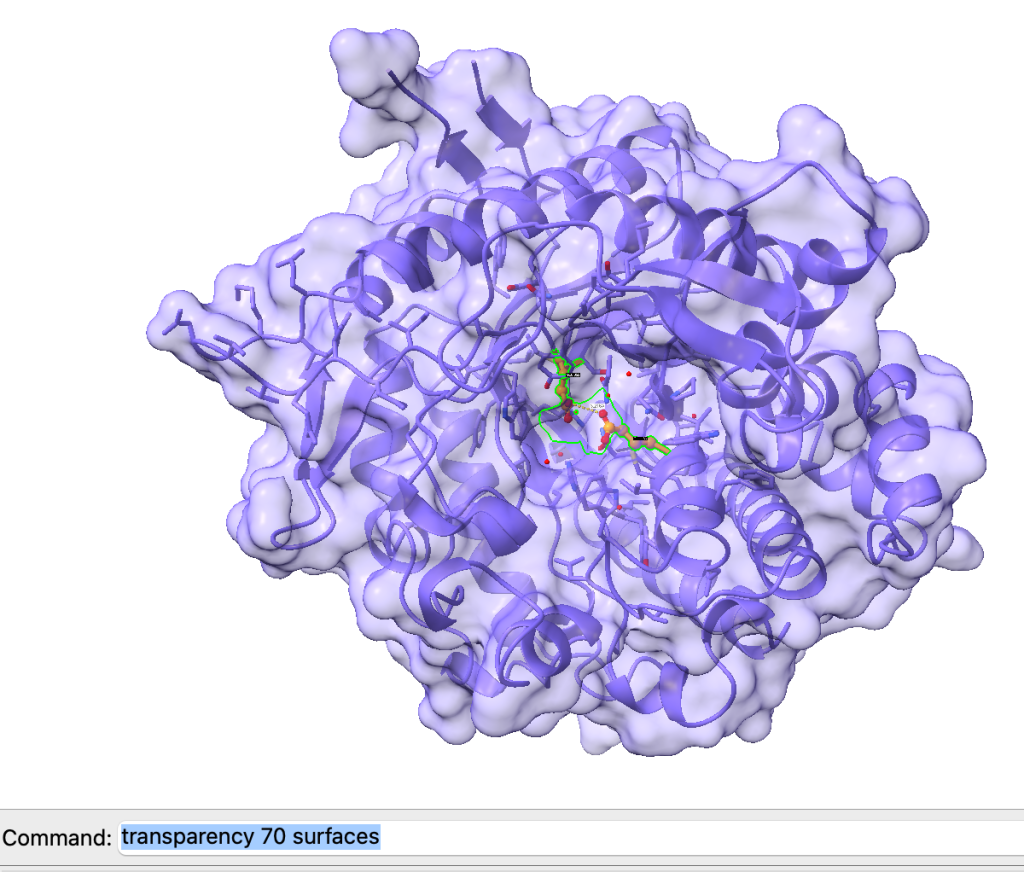
Para esconder as superfícies, use:
hide surfacesSelecionando um átomo pelo ID
Digite:
select #1@@serial_number=145429